
Bioinformatics Frameworks Part 1: Bash and Make
Background
Back when the first Solexa sequencers came out, there was a need for basic and functional tools for Next Generation Sequencing (NGS) data analysis. Because of the amount of data and the implications of the results, the data analysis was quite different compared to bioinformatics from before the introduction of short read sequencing. After each application of NGS has two or three alternative tools, ranging from RNA-Seq to structural genomic analysis, bioinformaticians started to string them together to make automated pipelines. These pipelines would transport and transform the output data from one tool to the input files for another tools, logging some of the results during the process.
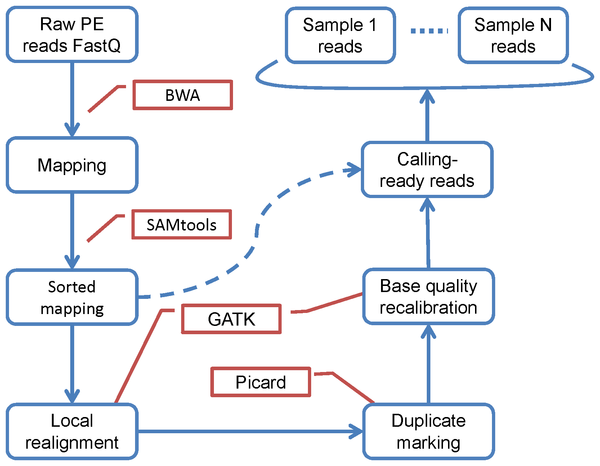
An example of a SNP calling pipeline workflow
At one point almost everyone working with DNA data had their own way of building these pipelines, with wildly varying levels usability, flexibly and reproducibility. More often than not, if a bioinformatician would leave a research team, the functionality of his/her pipelines would leave with him.
Frameworks
With that in mind, one of the biggest changes within the field of bioinformatics in recent years is the rise of the framework. Frameworks are the professionalization of these pipelines, constructed with software development concepts and data science in mind. These frameworks focus on flexibility, consistency, robustness, reproducibility, resource management, and if setup correctly, user friendliness. Most of them have ways of monitoring the jobs that you run and the manage the files that are involved. But again we see the same level of diversification: everyone seems to think that the wheel has to be reinvented, creating many different frameworks with a lot of overlap. With this series of blogs I will discuss a few that have crossed my path, and try to explain the essence of their design philosophy. It will not be a straight forward choice to pick the framework that suits your workflow best, but I hope these posts will help.
Prefacing: Bash
To start the series, why not go back to the very beginning. The oldest way of stringing commands, output file and tools together would be Unix Bash. Bash is basically the command line interface of Unix and more relevant; Linux. With Bash you can combine the commands you execute with some basic file checking and manipulation, most primitive pipelines would be build using Bash.
As an example for most of the frameworks, I’ll show basic SNP calling pipelines on NGS datasets. I’ll use real life examples, like this source:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 | #!/bin/bash # # initial example of a pipeline script bwa index reference.fa bwa aln -I -t 8 reference.fa s_1.txt > out.sai bwa samse reference.fa out.sai s_1.txt > out.sam samtools view -bSu out.sam | samtools sort - out.sorted java -Xmx1g -jar /apps/PICARD/1.95/MarkDuplicates.jar \ MAX_FILE_HANDLES_FOR_READ_ENDS_MAP=1000\ METRICS_FILE=out.metrics \ REMOVE_DUPLICATES=true \ ASSUME_SORTED=true \ VALIDATION_STRINGENCY=LENIENT \ INPUT=out.sorted.bam \ OUTPUT=out.dedupe.bam samtools index out.dedupe.bam samtools mpileup -uf reference.fa out.dedupe.bam | /apps/SAMTOOLS/0.1.19/bin/bcftools view -bvcg - > out.bcf |
Now, this is a functional pipeline: it runs from start to finish.
But it will only run if you have all the correct versions of the tools installed, the matching folder structure and PATH variables, the right files with the right names. It also doesn’t consider system resources, it’s not portable to other systems, it has no naming convention, it’s not easy to restarted if it fails for some reason. And if you don’t know some or all of the tools, parameters and files? Too bad.
You can make Bash scripts more elaborate, reproducible and fail-proof. But quite soon you’ll just be recreating your own version of the wheel. So all things considered, Bash is a scripting language, not a framework.
Make
What can be considered a framework is Make. Make is even older then Bash, in terms of computer science history Make is absolutely ancient. But it is still relevant: someone today would use Make to build a software from code in a Unix environment, basically to compile the executables from scratch. This functionality is starting to be replaced by things like yum and apt-get, but sometimes a tool is not on the binary repositories of your Linux distro, and you’ll use Make to install software.
Make is a prerequisite building system: it checks all the prerequisites of compiling a software tool before starting. You could say it takes the endpoint and works its way back. This works as well for compiling as it works for bioinformatics pipelines. An example can be found below:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 | .PHONY : bwa.index snpeff_download_db fastq bam bam.markdup strelka STRELKA_PATH = $(HOME)/usr/strelka/1.0.15/bin SNPEFF_PATH = $(HOME)/usr/snpeff/4.3 MUSEQ_PATH = $(HOME)/usr/museq/4.3.8/museq #---------- # Setup Reference Genome #---------- bwa.index : bwa index refs/GRCh37-lite.fa #---------- # Setup SnpEff #---------- SNPEFF_GENOME = GRCh37.75 # Download the SnpEff genome database snpeff_download_db : snpeff_$(SNPEFF_GENOME).timestamp snpeff_$(SNPEFF_GENOME).timestamp : java -Xmx4G -jar $(SNPEFF_PATH)/snpEff.jar \ download \ -c $(SNPEFF_PATH)/snpEff.config \ $(SNPEFF_GENOME) && touch $@ #---------- # Download Full Exome Data #---------- download : bam/gerald_C1TD1ACXX_7_CGATGT.bam bam/gerald_C1TD1ACXX_7_ATCACG.bam # Exome Normal bam/gerald_C1TD1ACXX_7_CGATGT.bam : mkdir -p $(@D); \ wget -p $(@D) https://xfer.genome.wustl.edu/gxfer1/project/gms/testdata/bams/hcc1395/gerald_C1TD1ACXX_7_CGATGT.bam # Exome Tumor bam/gerald_C1TD1ACXX_7_ATCACG.bam : mkdir -p $(@D); \ wget -p $(@D) https://xfer.genome.wustl.edu/gxfer1/project/gms/testdata/bams/hcc1395/gerald_C1TD1ACXX_7_ATCACG.bam #--------- # Fastq Extraction #--------- fastq : fastq/gerald_C1TD1ACXX_7_CGATGT_R1.fastq \ fastq/gerald_C1TD1ACXX_7_CGATGT_R2.fastq \ fastq/gerald_C1TD1ACXX_7_ATCACG_R1.fastq \ fastq/gerald_C1TD1ACXX_7_ATCACG_R2.fastq fastq/%_R1.fastq fastq/%_R2.fastq : bam/%.bam picard SamToFastq \ INPUT=$< \ FASTQ=fastq/$*_R1.fastq \ SECOND_END_FASTQ=fastq/$*_R2.fastq #---------- # Sequence Alignment using BWA #---------- sai/%.sai: fastq/%.fastq mkdir -p $(@D); \ bwa aln -t 4 refs/GRCh37-lite.fa $< > $@ 2> log/$(@F).log bam : bam/HCC1395_exome_normal.bam bam/HCC1395_exome_tumour.bam bam/HCC1395_exome_normal.bam : sai/gerald_C1TD1ACXX_7_CGATGT_R1.sai sai/gerald_C1TD1ACXX_7_CGATGT_R2.sai fastq/gerald_C1TD1ACXX_7_CGATGT_R1.fastq fastq/gerald_C1TD1ACXX_7_CGATGT_R2.fastq bwa sampe refs/GRCh37-lite.fa $^ | samtools view -bh > $@ bam/HCC1395_exome_tumour.bam : sai/gerald_C1TD1ACXX_7_ATCACG_R1.sai sai/gerald_C1TD1ACXX_7_ATCACG_R2.sai fastq/gerald_C1TD1ACXX_7_ATCACG_R1.fastq fastq/gerald_C1TD1ACXX_7_ATCACG_R2.fastq bwa sampe refs/GRCh37-lite.fa $^ | samtools view -bh > $@ #---------- # Post-Processing of BAM Files #---------- bam.sort.markdup : bam/HCC1395_exome_normal.sort.markdup.bam bam/HCC1395_exome_tumour.sort.markdup.bam bam/%.sort.bam : bam/%.bam picard SortSam \ I=$< \ O=$@ \ SORT_ORDER=coordinate \ VALIDATION_STRINGENCY=LENIENT bam/%.markdup.bam : bam/%.bam mkdir -p bam/markdup_stats; \ picard MarkDuplicates \ I=$< \ O=$@ \ M=bam/markdup_stats/$*_marked_dup_metrics.txt \ VALIDATION_STRINGENCY=LENIENT # Generate Samtools Index %.bam.bai : %.bam samtools index $< #---------- # Filter Bam for Only 1MB region of Chromosome 17 #---------- filtered.bam : \ bam/HCC1395_exome_normal.sort.markdup.17.7MB-8MB.bam \ bam/HCC1395_exome_normal.sort.markdup.17.7MB-8MB.bam.bai \ bam/HCC1395_exome_tumour.sort.markdup.17.7MB-8MB.bam \ bam/HCC1395_exome_tumour.sort.markdup.17.7MB-8MB.bam.bai bam/HCC1395_exome_normal.sort.markdup.17.7MB-8MB.bam : bam/HCC1395_exome_normal.sort.markdup.bam samtools view -b $< 17:7000000-8000000 > $@ bam/HCC1395_exome_tumour.sort.markdup.17.7MB-8MB.bam : bam/HCC1395_exome_tumour.sort.markdup.bam samtools view -b $< 17:7000000-8000000 > $@ #---------- # Variant Calling #---------- # Run MutationSeq on Full Exome museq/results/HCC1395_exome_tumour_normal.vcf : bam/HCC1395_exome_normal.sort.markdup.bam bam/HCC1395_exome_tumour.sort.markdup.bam bam/HCC1395_exome_normal.sort.markdup.bam.bai bam/HCC1395_exome_tumour.sort.markdup.bam.bai mkdir -p $(@D); \ python $(MUSEQ_PATH)/classify.py \ normal:$< \ tumour:$(word 2,$^) \ reference:refs/GRCh37-lite.fa \ model:$(MUSEQ_PATH)/models_anaconda/model_v4.1.2_anaconda_sk_0.13.1.npz \ -c $(MUSEQ_PATH)/metadata.config \ -o $@ |
This code is located in the Makefile, that’s the actual file name recognized by Unix itself. The coding structure and rules are very rigid and ancient, it does not compare to something like Python or R. Using Make means that you will be debugging it a lot, and many conventions are not logical anymore. For example when using filename variables that carry over; the difference between $^ and $< can be overlooked easily but have a big impact if used incorrectly. So a big “no” when it comes to user friendliness.
But you can interrupt it and it is robust: files that are created in a previous run are taken into account. You can assign instances to it, so you can run parallel versions on multiple cores and even other computer nodes. It has built in naming conventions as seen in the example, it is reproducible and reasonably portable.
Considering all these factors, you could say that Make meets the bare bone requirements of a framework.
Conclusion
So that the way things started out in the field of bioinformatics pipeline frameworks. It is good to keep these examples in mind when comparing and judging actual frameworks. In the next parts I will be looking at more modern approaches. Stay tuned!
