
Bioinformatics Frameworks Part 4: Nextflow
Introduction
One of the biggest changes within the field of bioinformatics is the rise of the framework. Frameworks are the professionalization of analysis pipelines, constructed with software development and data science in mind. With this series of blogs I will discuss a few that have crossed my path, and try to explain the essence of their design philosophy. In part four, I will focus on Nextflow, the framework build for scaling.
Local vs. Scaled
In the previous post in this series, I discussed SnakeMake, the Python based update for bioinformatics pipelines that were made using the archaic Unix Make system. The example commands that I showed in that posed were performed as local executions; SnakeMake loads the files, runs the commands and manages the resources on a single computer with local storage. This is fine and dandy when dealing with either relatively small datasets or spacious deadlines. But with the current deluge of data generated by the Illumina HiSeq or, even worse, the NovaSeq systems, local systems will not suffice if you want to provide timely data analysis results.
The move from local execution to server cluster execution (also called scaled up execution) has been implemented long ago for many use cases. For that purpose, several HPC (High Perfomance Computing) computational frameworks already exist. With examples like Hadoop from Apache and qsub from the Oracle/Sun Grid Engine, there are several ways for scheduling and executing computational jobs on clusters or cloud grids. This also applies to SnakeMake and even Make; newer versions of SnakeMake supports qsub and scaled containerized execution via Kubernetes. Make has a qMake, which I would call the most basic job scheduler of them all, but hey, it works.
However, there is one framework that was built from the start with all the best framework practices in mind. By that I mean the design principles that I mentioned in previous posts:
- Both local and scaled execution
- Docker or containerized commands
- Fully reproducible
- Crash/failure robustness
- Versioning for pipelines, software and files
- Software package management
- And lastly, resource management
This framework is called…
![]()
Nextflow
This framework is not advertised to be developed for bioinformatics in mind, but there are many examples on the main site and github that concern bioinformatics software. Furthermore, it has sizeable traction amongst bioinformaticians, with several Core Sequencing centers that converted their production to Nextflow.
By now, I’ve discussed the structure of bioinformatics pipelines several times, so I won’t repeat myself again. I’ll give an example of a Nextflow-based RNA-seq pipeline below, it’s all quite straight forward.
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 | #!/usr/bin/env nextflow /* * Defines pipeline parameters in order to specify the refence genomes * and read pairs by using the command line options */ params.reads = "$baseDir/data/ggal/*_{1,2}.fq" params.genome = "$baseDir/data/ggal/ggal_1_48850000_49020000.Ggal71.500bpflank.fa" /* * The reference genome file */ genome_file = file(params.genome) /* * Creates the `read_pairs` channel that emits for each read-pair a tuple containing * three elements: the pair ID, the first read-pair file and the second read-pair file */ Channel .fromFilePairs( params.reads ) .ifEmpty { error "Cannot find any reads matching: ${params.reads}" } .set { read_pairs } /* * Step 1. Builds the genome index required by the mapping process */ process buildIndex { input: file genome from genome_file output: file 'genome.index*' into genome_index """ bowtie2-build ${genome} genome.index """ } /* * Step 2. Maps each read-pair by using Tophat2 mapper tool */ process mapping { input: file genome from genome_file file index from genome_index set pair_id, file(reads) from read_pairs output: set pair_id, "tophat_out/accepted_hits.bam" into bam_files """ tophat2 genome.index ${reads} """ } /* * Step 3. Assembles the transcript by using the "cufflinks" * and publish the transcript output files into the `results` folder */ process makeTranscript { publishDir "results" input: set pair_id, bam_file from bam_files output: set pair_id, 'transcripts.gtf' into transcripts """ cufflinks ${bam_file} """ } |
Nextflow is polyglot: where SnakeMake requires a background in Python coding and Make requires background in medieval coding voodoo and bucket loads of patience, Nextflow works with many types of programming languages.
Another difference with SnakeMake is how it stores the files; for each step/command in the pipeline a different folder is created in the “work” directory, with a random hexadecimal as name. Each of these folders contains all the files that are required for execution, and the input files are symlinked from the previous folder/step. The output files are created, and serve as the basis for symlinks in the next folder/step. Its not easy for humans to find intermediary files, but its great for reproducibility and crash recovery since all steps are isolated.
The framework supports many of the same features as SnakeMake, like pipeline visualisations, automated reporting and tracing (note the random pipeline execution name; angry_babbage):
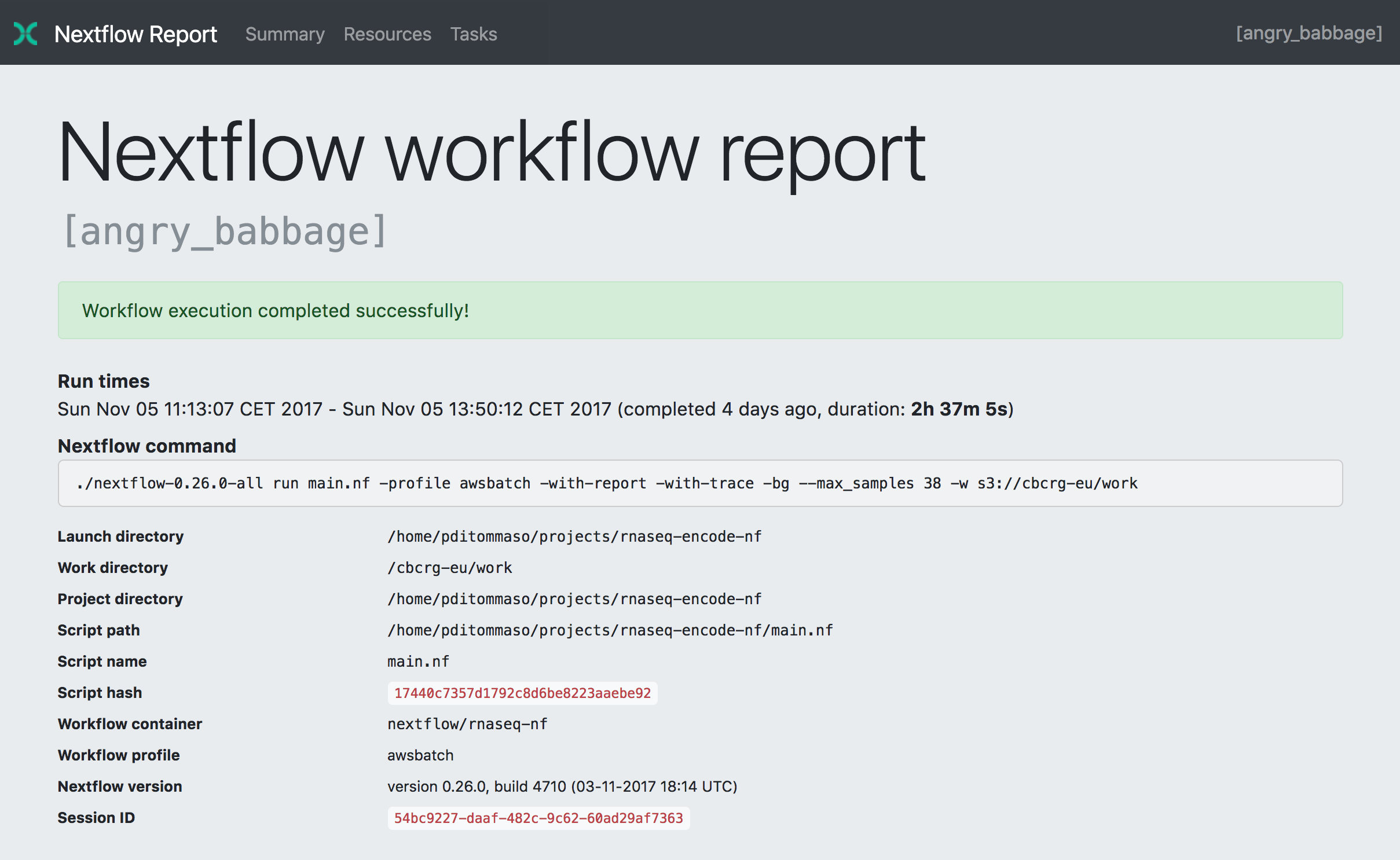

The implementation of Docker, Package management and Github are native and almost mandatory, much more than SnakeMake. Although a bit strict, this does have the benefit that after installation, you can run the example RNA-Seq pipeline directly from the official Github, without any configuration. Like this:
1 | nextflow run rnatoy -with-docker |
Conclusion
Granted, on the surface and when skimming through the specification list, Nextflow looks a lot like SnakeMake. SnakeMake also passes many of the requirements that I iterated earlier in this post, included scaled up executions. But a lot of these features were added in later versions of SnakeMake, so it seems that it was initially constructed as a Make replacement and later as a fully-fledged future-proof framework (the three F’s). For Nextflow, the three F’s were implemented from the get go. For example, Nextflow supports Kubernetes just like SnakeMake. But it also natively supports Amazon Cloud and AWS Batch, unlike SnakeMake. After working with Nextflow, I do feel that this workflow language is the go-to framework for large scale bioinformatics deployment.
